
7 días en 7 noticias, 1 cifra y 1 frase:...
Leer más
SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex
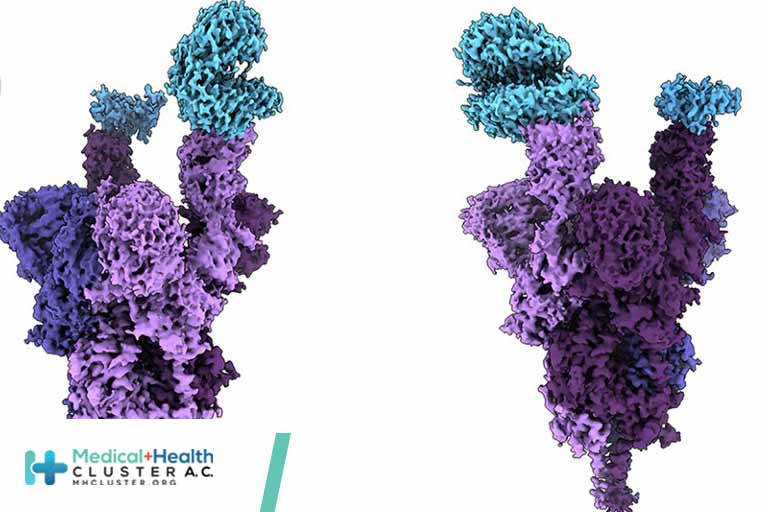
Antibody-evading Omicron keeps function
The Omicron variant of severe acute respiratory syndrome coronavirus 2 was reported in November 2021 and was quickly identified as a variant of concern because of its rapid spread. Relative to the original Wuhan-Hu-1 strain, this variant has 37 mutations in the spike protein that is responsible for binding and entry into host cells. Fifteen mutations are in the receptor-binding domain, which binds the host angiotensin-converting enzyme 2 (ACE2) receptor and is also the target of many neutralizing antibodies. Mannar et al. report a structure of the Omicron variant spike protein bound to human ACE2. The structure rationalizes the evasion of antibodies elicited by previous vaccination or infection and shows how mutations that weaken ACE2 binding are compensated for by mutations that make new interactions. —VV
Abstract
The newly reported Omicron variant is poised to replace Delta as the most prevalent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant across the world. Cryo–electron microscopy (cryo-EM) structural analysis of the Omicron variant spike protein in complex with human angiotensin-converting enzyme 2 (ACE2) reveals new salt bridges and hydrogen bonds formed by mutated residues arginine-493, serine-496, and arginine-498 in the receptor binding domain with ACE2. These interactions appear to compensate for other Omicron mutations such as the substitution of asparagine for lysine at position 417 (K417N) that are known to reduce ACE2 binding affinity, resulting in similar biochemical ACE2 binding affinities for the Delta and Omicron variants. Neutralization assays show that pseudoviruses that display the Omicron spike protein exhibit increased antibody evasion. The increase in antibody evasion and the retention of strong interactions at the ACE2 interface thus represent important molecular features that likely contribute to the rapid spread of the Omicron variant.
The Omicron (B.1.1.529) variant of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), first reported in November 2021, was quickly identified as a variant of concern with the potential to spread rapidly across the world. This concern is heightened because the Omicron variant is now circulating even among doubly vaccinated individuals. SARS-CoV-2 relies on a trimeric spike protein for host cell entry via recognition of the angiotensin-converting enzyme 2 (ACE2) receptor. The Omicron variant spike protein has 37 mutations, as compared to 12 mutations in the Gamma variant spike protein, which was previously the variant with the greatest number of spike protein mutations (1). Understanding the consequences of these mutations for ACE2 receptor binding and neutralizing antibody evasion is important in guiding the development of effective therapeutics to limit the spread of the Omicron variant and related variants.The spike protein comprises two domains: the S1 domain, which contains the receptor binding domain (RBD), and the S2 domain, which is responsible for membrane fusion. The Omicron variant has 37 mutations (Fig. 1A) in the spike protein relative to the initial Wuhan-Hu-1 strain, with 15 of them present in the RBD (1). The RBD mediates attachment to human cells through the ACE2 receptor and is the primary target of neutralizing antibodies (2, 3). The Delta variant, which was the predominant SARS-CoV-2 lineage until the emergence of Omicron, has seven mutations in the spike protein relative to the Wuhan-Hu-1 strain, with two mutations falling within its RBD. Of the Delta spike protein mutations, two [T478K (Thr478→Lys) in the RBD and D614G (Asp614→Gly) at the C terminus of S1] are shared with the Omicron strain. Analysis of the sequence of the Omicron genome suggests that it is not derived from any of the variants circulating at present and may have a different origin (4).
Cryo–electron microscopy (cryo-EM) structural analysis of the Omicron spike protein ectodomain shows that the overall organization of the trimer is similar to that observed for the ancestral strain (5–7) and all earlier variants (8–10) (Fig. 1B and table S1). The RBD in one of the protomers (protomer 1) is well-resolved and is in the “down” position, whereas the other two RBDs are less well-resolved because they are flexible relative to the rest of the spike protein polypeptide. Similarly, the amino terminal domain (NTD) is poorly resolved, reflecting the dynamic and flexible nature of this domain. The mutations in the Omicron variant spike protein are distributed both on the surface and the interior of the spike protein (Fig. 1C), including the NTD and RBD regions. The mutations in the RBD are predominantly distributed on one face of the domain (Fig. 1D), which spans regions that bind ACE2 as well as those that form epitopes for numerous neutralizing antibodies (11).
The Omicron variant shares RBD mutations with previous variants of concern [K417N (Lys417→Asn), T478K, and N501Y (Asn501→Tyr)]. The N501Y and K417N mutations impart increased and decreased ACE2 binding affinities, respectively (8, 12–16). These mutational effects preserve the same general impact on ACE2 affinity when present in isolation or in combination with other RBD mutations (12). However, the Omicron RBD contains additional mutations, most of which have been shown to decrease receptor binding in a high-throughput assay (table S2) (17), with the exception of G339D (Gly339→Asp), N440K (Asn440→Lys), S447N (Ser447→Asn), and Q498R (Gln498→Arg) (17, 18). To measure the impact of Omicron spike protein mutations on human ACE2 binding affinity, we performed surface plasmon resonance (SPR) studies and compared the resulting apparent binding affinities (KD,app) to wild-type and Delta spike proteins (Fig. 2). “Wild type” is used in this work to refer to the ancestral Wuhan-Hu-1 strain with the addition of the D614G mutation. Although the Omicron spike protein exhibits a measurable increase in apparent affinity for ACE2 relative to the wild-type spike protein [in agreement with a recent preprint (19)], the apparent ACE2 affinity is similar for both the Delta and Omicron variants (Fig. 2D). Despite harboring several RBD mutations that decrease ACE2 binding (fig. S2) (12, 16, 17), the preservation of overall ACE2 binding affinity for the Omicron spike protein suggests there are compensatory mutations that restore higher affinity for ACE2. Such mutational effects should be possible to visualize in a high-resolution structure of the spike protein–ACE2 complex.

FIG. 2. SPR analysis of the wild-type, Delta, and Omicron spike protein affinities for human ACE2.
(A to C) Representative traces of single-cycle kinetic analyses of spike protein–ACE2 binding. The raw data (black) is fit (red) to a model using a 1:1 binding stoichiometry from which apparent dissociation constants were derived. The curves were obtained by injecting 6.25, 31.25, 62.5, 125, and 250 nM of each spike protein in successive cycles. RU, response units; WT, wild type. (D) Quantitation of apparent dissociation constants (KD,app) for the wild-type, Delta, and Omicron spike protein–ACE2 interactions. The standard deviation obtained from at least three technical replicates is shown. Horizontal dotted lines are plotted for mutants carrying only K417N (top) or N501Y and E484K (Glu484→Lys; bottom) mutations to demonstrate the range of this assay (see fig. S2 for binding data). A Tukey’s multiple comparisons test was performed on the wild-type, Delta, and Omicron binding affinities (*P ≤ 0.05; ns, not significant). A table highlighting the fold changes in KD,app for the Delta and Omicron spike protein–ACE2 interactions relative to wild type is shown.
Cryo-EM structural analysis of the human ACE2–Omicron spike protein complex shows strong density for ACE2 bound to the RBD of one of the protomers in the “up” position (Fig. 3A and table S1). Weaker density is observed for a second bound ACE2, suggesting partial occupancy of a second RBD under our experimental conditions. We focus on the structure of the ACE2–spike protein interface in the most strongly bound ACE2 molecule. Focused refinement of the RBD-ACE2 region resulted in a density map with a resolution of 2.66 Å at the spike protein–ACE2 interface (Fig. 3B), allowing the visualization of side chains involved in the interface (Fig. 3C). In Fig. 3, D to F, we compare the key interactions at this interface in the Omicron variant with corresponding interactions that we have recently reported for the Delta variant (20). In the Delta variant–ACE2 complex, there are hydrogen bonds formed by residues Q493 and Q498 on the spike protein with residues E35 (E, Glu) and Q42, respectively, on ACE2 (Fig. 3D). In the Omicron variant, three mutations are observed in this stretch: Q493R (Gln493→Arg), G496S (Gly496→Ser), and Q498R. Residue R493 replaces the hydrogen bond to ACE2 residue E35 with a new salt bridge, whereas residue R498 forms a new salt bridge with ACE2 residue D38 while maintaining a hydrogen bond interaction with ACE2 residue Q42. RBD residue S496 adds a new interaction at the interface by forming a hydrogen bond with ACE2 residue K353 (Fig. 3D). Additionally, the mutated residue Y501 in the Omicron RBD makes π-stacking interactions with Y41 in ACE2, as previously seen in the Alpha (B.1.1.7), Beta (B.1.351), and Gamma (P.1) variants (8, 12), whereas mutated residue H505 (H, His) is not hydrogen-bonded to E37 in ACE2, in contrast to what we reported previously for the Y505 residue (Fig. 3E) (20).
Créditos: Comité científico Covid


